Usher Syndrome Research
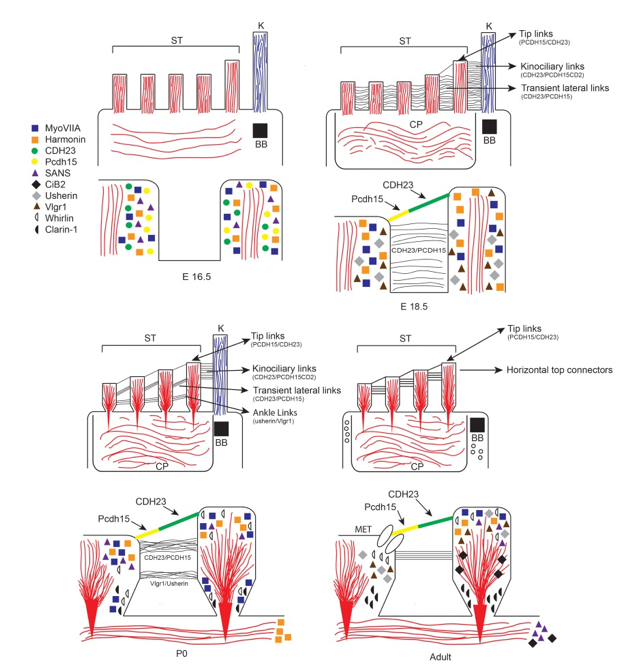
Usher Syndrome is a genetically heterogeneous disorder characterized by congenital deafness associated with delayed onset and progressive retinitis pigmentosa. There are 11 genes encoding a much larger number of protein variants including a non-conventional myosin, scaffold proteins, a g-protein coupled receptor, a calcium integrin binding protein, and cell adhesion proteins. When mutated, cochlear hair cells develop abnormal stereocilia that are splayed, abnormal lengths, and often oriented abnormally within the cuticular plate, indicating a defect in planar cell polarity.These defects are evident by embryonic day 18 in the mouse, long before the onset of hearing. In mature hair cells, a number of Usher proteins are critical components tip links of the stereocilia and of the mechanotransduction channel apparatus which gates the depolarization of the hair cells in response to noise.
All of the Usher proteins are also expressed in the photoreceptors. We have identified a role for these proteins in regulating the light dependent translocation of phototransduction proteins, and find that many of the Usher proteins localize to the transition zone or basal body aspect of the connecting cilia, which infers a possible function in regulating protein trafficking between the inner and outer segments.
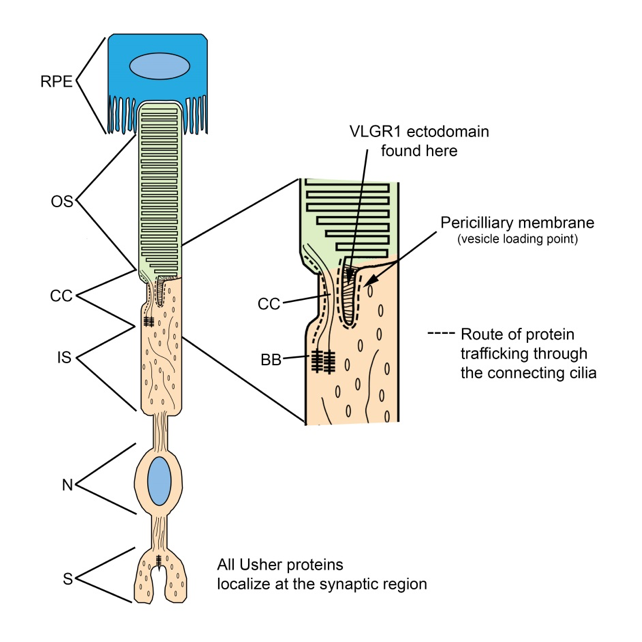 There are many interactions that have been demonstrated for the Usher proteins, however the complexity of this system has made progress towards identification of the specific functions of these complexes somewhat elusive. Most Usher proteins localize to the synapses of hair cells and photoreceptors as well, where they may play a role in synaptic maturation.
There are many interactions that have been demonstrated for the Usher proteins, however the complexity of this system has made progress towards identification of the specific functions of these complexes somewhat elusive. Most Usher proteins localize to the synapses of hair cells and photoreceptors as well, where they may play a role in synaptic maturation.
The
Functional Genetics Laboratory with
Dr. Marisa Zallocchi in the
Center for Sensory Neuroscience was created to implement a high throughput forward genetic approach for dissecting the function of Usher protein complexes using the zebrafish model system.

